Modulation of Signal Transduction
Growth factors acting through receptor tyrosine kinases (RTKs) of ERBB family, integrate a network of multiple inputs, such as ligands of cell-surface receptors; and produce multiple outputs, from gene expression and cellular activities, including motility, proliferation and death. With the “omics era”, the complexity of both intra e inter cellular interactions become manifest, thus our mission is trying to understand the special laws of nature modelling biological systems.
Transcription networks are designed with specific timescales. In response to a stimuli signal-transduction networks operate immediately, by transmitting the input to several Transcription Factors and once on the DNA, they are engaged in the production of target genes.
Steroid hormones activating nuclear localized receptors, are able to bypass the transduction networks, thus the timing of their activities result faster and relatively simple. Nevertheless, in a living organism, these two systems interact massively, with major consequences for body homeostasis and cell activities.
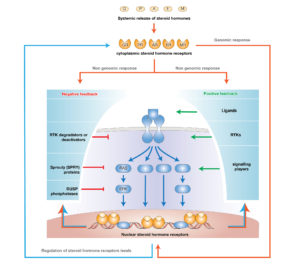
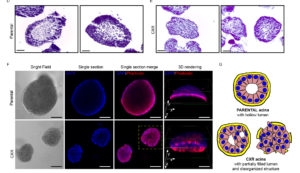
This line of research focuses on the transcriptional modifications resulting from steroid hormones and growth factors interaction. 
We previously showed that capturing epigenetic mechanisms and transcriptional control of several feedback regulatory loops, steroid hormones (GCs) gained the ability to robustly inhibit EGF induced signals leading to cell migration. Specifically, GCs enhance auto-inhibitors of EGF signaling and inhibit expression of auto-stimulators of the same pathway.
We are studying the mechanisms driving this interaction in order to optimize the response to EGFR target therapy, in several biological backgrounds, either mammary epithelial tissues and gastrointestinal tissues.
Modulation of Drug Response
Resistance to EGFR blockade may have a genetic basis as well as activation of downstream or parallel signaling pathways that substitute for EGFR inhibition. This process relies on plastic, reversible traits induced by drug pressure, such as compensatory activation of biochemical feedback circuits and transcriptional modifications.
In this project, we aim to explore the plastic phenotype of cellular adaptation to prolonged drug treatment.
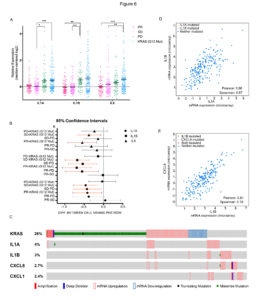
We found that resistance to EGFR targeting drugs results in the up-regulation of a signature of inflammatory cytokines, namely IL1A, IL1B and IL8. The association between reduced sensitivity to anti-EGFR antibodies and increased expression of this inflammatory signature was confirmed in patients’ data and specimens.
We employ monolayer and 3D-spheroid growing cells to decipher the mechanisms by which an inflammatory microenvironment might impair cetuximab efficacy in patients.